

Cancer & Gene Regulation
Gene regulation is frequently dysregulated in cancer. During cancer initiation, transcription factors can exhibit somatic mutations including point mutations in regulatory domains or undergo copy number variations due to genetic deletion. The essential chromatin organising protein and tumour suppressor gene CTCF is central to our research focus. Mutations in CTCF occur frequently in endometrial and breast cancers, as well as leukemia. We are functionally characterising mutations in CTCF and their impact on DNA binding, protein-protein interactions, chromatin organisation and cell growth characteristics. More broadly, we are also examining the roles of other tumour suppressor genes (e.g. MGA) and chromatin remodelling complexes (e.g. NuRD) in normal biology and cancer. Our research is revealing new insights into the molecular pathogenesis of cancer initiation and progression.
Projects
Molecular modeling of cancer-associated mutations in tumour suppressor genes

Molecular docking of somatic CTCF ZF mutations with DNA
We are currently modelling ‘hotspot’ mutations on published CTCF ZF protein structures to assess the impact of those mutations. In addition, we will develop homology models for the ZF domain of CTCF-Like protein (CTCFL, also known as BORIS). CTCFL which shares 80% homology with CTCF within its ZF domain, and which is phenotypically similar to CTCF is aberrantly expressed in more than half of all cancers. Both factors have overlapping and unique DNA binding characteristics. We will examine which residues are critical for binding of CTCF and CTCFL to the same DNA target sites and which ZF residues confer DNA binding specificity. This will provide important insight into the sibling rivalry that exists between CTCF and CTCFL in normal biology and cancer.
Find out more: Bailey et al, Cellular & Molecular Life Sciences 2021
Dissecting the role of ZNF185 in CTCF-mutant endometrial cancer
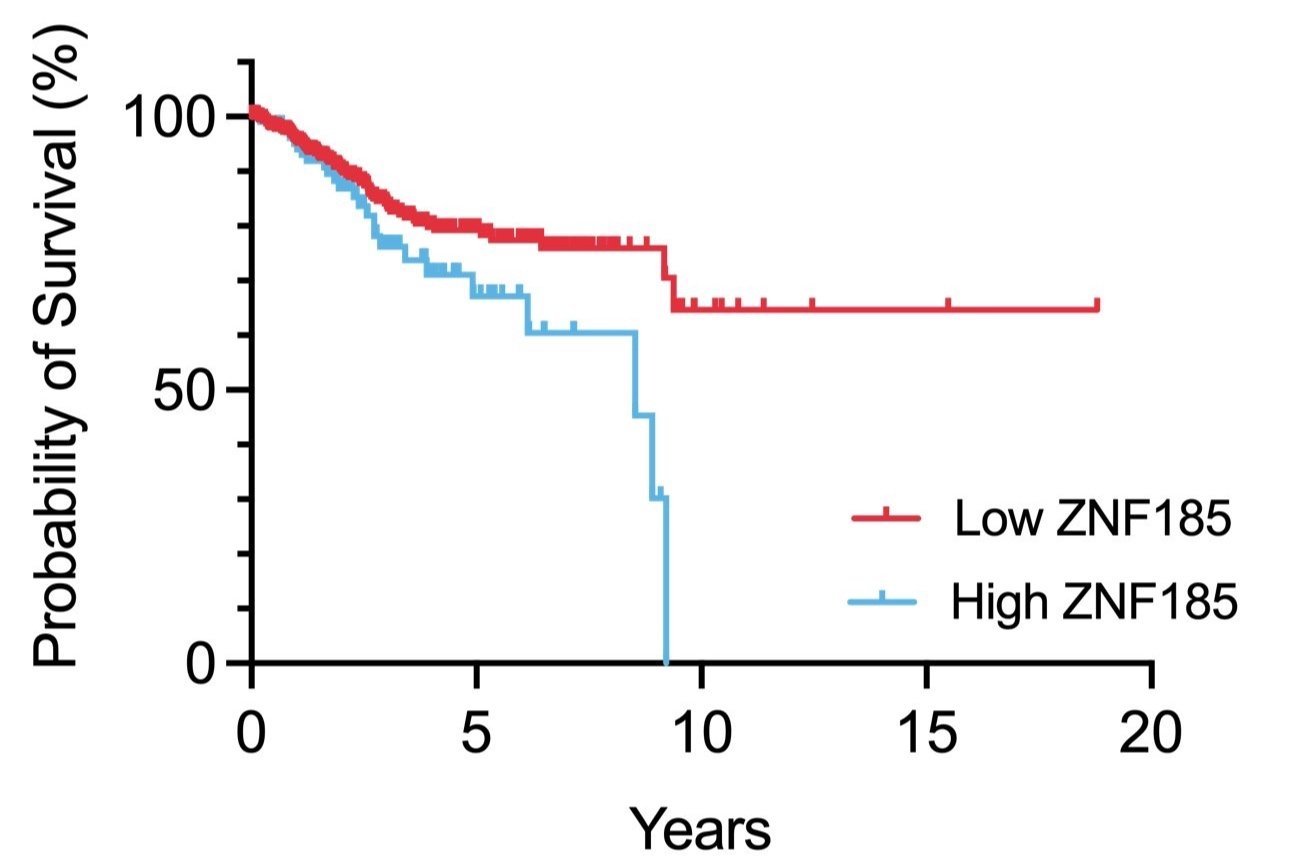
Survival analysis of ZNF185 expression in endometrial cancer
Normal gene expression is frequently dysregulated in cancer. The tumour suppressor CTCF, an essential regulator of transcription and chromatin architecture, is frequently mutated in cancer. The molecular pathogenesis underlying CTCF mutation in hormone-responsive cancers in women is poorly understood. This project focuses on ZNF185, an actin cytoskeleton remodeling protein which is induced upon CTCF mutation in endometrial cancer. How ZNF185 expression increases cell proliferation and alters cell polarity will be examined using overexpression and knockdown/knockout studies. Transcriptomics, proteomics and protein interaction studies will be used to identify key determinants in the molecular pathophysiology of endometrial cancers.
Find out more: Marshall et al, Oncogene 2017
Understanding the role of CTCF genetic deletion in aggressive endometrial cancer

CTCF shRNA knockdown in KLE endometrial cancer spheroids
Our team has discovered that CTCF is genetically deleted at high rates in the most aggressive and deadly types of endometrial cancer. CTCF deletion predominantly occurs in the Type II serous subtype of endometrial cancer and is associated with poorer overall survival in patients with serous tumours. Our culturing of endometrial cancer cell-lines as 3D spheroids has shown that a functional consequence of CTCF deletion in results in a loss of cell polarity – an early event in endometrial cancer pathology. We are examining those genes and biochemical pathways that are dysregulated in CTCF mutant endometrial cancers. This will give important insights into early pathophysiological events underlying endometrial cancer.
Find out more: Marshall et al, Oncogene 2017
The role of MGA mutation in chronic lymphocytic leukaemia (CLL)

Homology modelling of the MGA/MAX BHLHzip heterodimer interacting with DNA
CLL is a slow developing cancer affecting B cells. Genetic mutations acquired in these B cells result in their transformation into cancerous cells that can live longer and grow faster than normal B cells. Similar to many blood cancers, genetic alterations in CLL can be heterogeneous. However, recent reports have identified the gene encoding the transcription factor Max Gene Associated (MGA) to be recurrently deleted or mutated in CLL. Our hypothesis is that genetic inactivation of MGA promotes chronic lymphocytic leukaemia disease progression. We will test this hypothesis by analysing how acquired genetic lesions in MGA alter the proliferation, differentiation and survival of CLL cells and contribute to cellular transformation.